Solvent
Defining solvent molecule
Solvent molecules can be easily added in analogous way to the main molecule. First the solvent molecule is defined as a PyQchem Structure object.
Example of water:
solvent = Structure(coordinates=[[0.000000, 0.000000, 0.000000],
[0.758602, 0.000000, 0.504284],
[0.758602, 0.000000, -0.504284]
],
symbols=['O', 'H', 'H'])
then the solvent molecule is added during the calculation setup:
calc = GromOrg(structure,
params=gmx_params, # MDP parms
box=[30, 30, 30], # unitcell a, b, c in angstrom
supercell=[1, 1, 1], # size of supercell
solvent=solvent, # solvent molecule
solvent_scale=0.57, # solvent scale parameter
)
To adjust the density of the solvent molecule, the solvent_scale parameter can be used.
This correspond to the -scale option in the gmx solvate command.
(https://manual.gromacs.org/current/onlinehelp/gmx-solvate.html?highlight=solvate)
Extracting molecular cluster from trajectory
To extract the molecules from the trajectory mdtraj_to_pyqchem can be used as previously.
However, when including the solvent it becomes convenient to extract not only the main molecule
but also some of closest solvent molecules. For this purpose get_cluster function can be used.
cluster = get_cluster(trajectory, iframe, ires, cutoff=5.0, center=True)
where iframe is the frame number, ires is the residue number and cutoff is the distance cutoff.
Also, center is a boolean parameter that can be used to center the molecule on the origin. The return
value is a PyQchem Structure object that contains the molecule corresponding to ires and the molecules
surrounding it at less than cutoff distance.
Note
Notice that in the trajectory the first residue indices always correspond to the main molecule while the
others are solvent molecules. Using the value defined in supercell the user can figure out how many
main molecules are present in the trajectory.
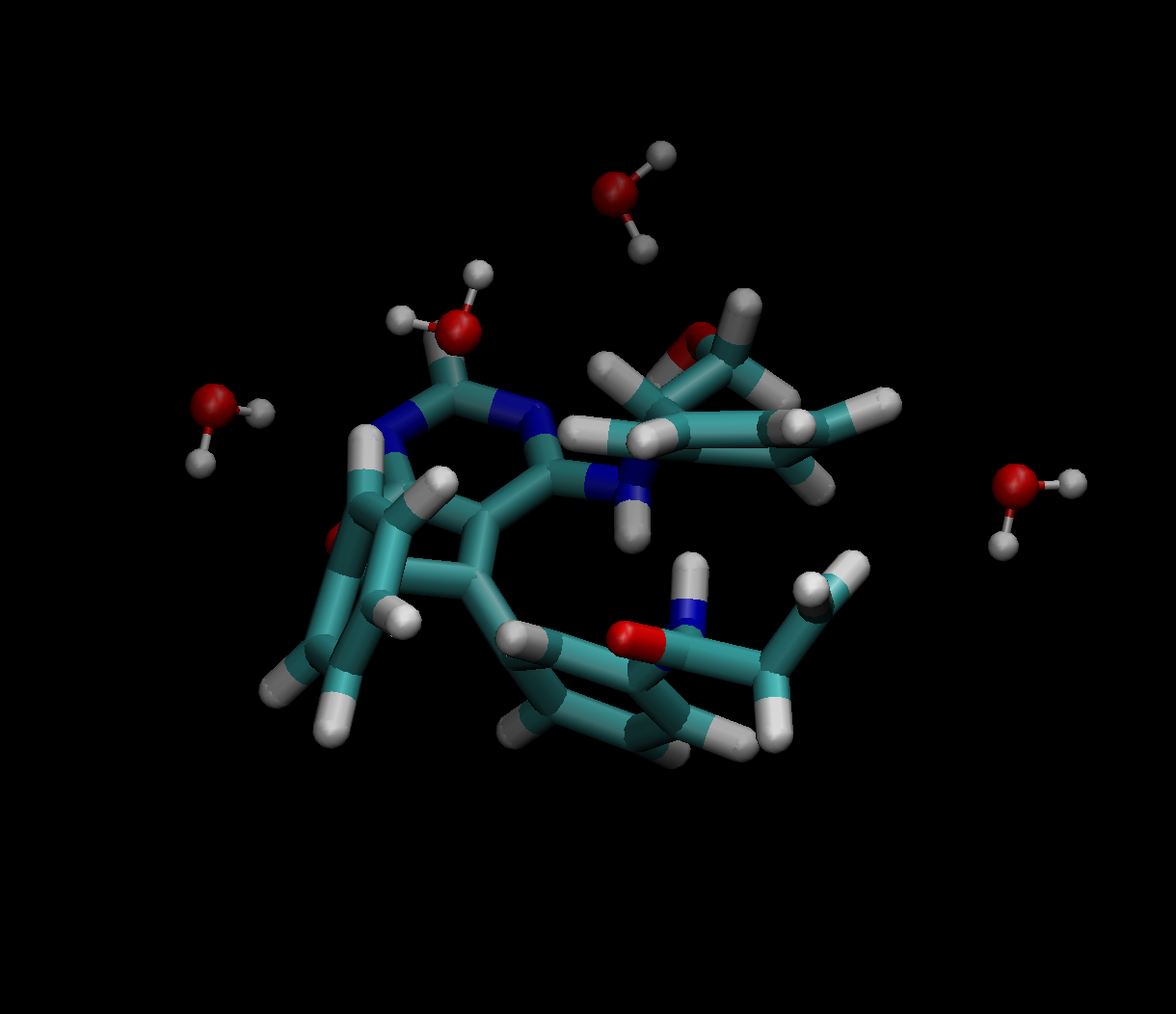
Simple example of the Structure (main mol + solvent) obtained using get_cluster function.